Ion Mobility Mass Spectrometry Uncovers the Impact of the Patterning of Oppositely Charged Residues on the Conformational Distributions of Intrinsically Disordered Proteins
Abstract
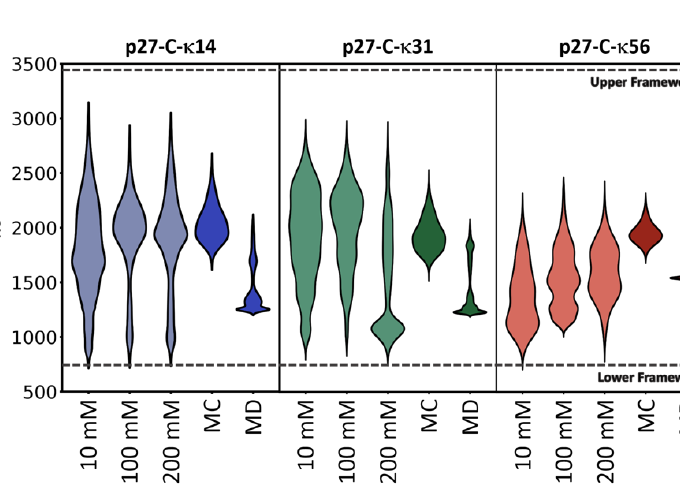
The global dimensions and amplitudes of conformational fluctuations of intrinsically disordered proteins are governed, in part, by the linear segregation versus clustering of oppositely charged residues within the primary sequence. Ion Mobility-Mass Spectrometry (IM-MS) affords unique advantages for probing the conformational consequences of the linear patterning of oppositely charged residues because it measures and separates proteins electrosprayed from solution on the basis of charge and shape. Here, we use IM-MS to measure the conformational consequences of charge patterning on the C-terminal intrinsically disordered region (p27 IDR) of the cell cycle inhibitory protein p27Kip1. We report the range of charge states and accompanying collisional cross section distributions for wild-type p27 IDR and two variants with identical amino acid compositions, κ14 and κ56, distinguished by the extent of linear mixing versus segregation of oppositely charged residues. Wild-type p27 IDR (κ31) and κ14 where the oppositely charged residues are more evenly distributed, exhibit a broad distribution of charge states. This is concordant with high degrees of conformational heterogeneity in solution. By contrast, κ56 with linear segregation of oppositely charged residues, leads to limited conformational heterogeneity and a narrow distribution of charged states. Molecular dynamics simulations demonstrate that the interplay between chain solvation and intra-chain interactions (self-solvation) leads to conformational distributions that are modulated by salt concentration, with the wild-type sequence showing the most sensitivity to changes in salt concentration. These results suggest that the charge patterning within the wild-type p27 IDR may be optimized to sample both highly solvated and self-solvated conformational states.